
Leica INM100 Optical Microscope Operation Manual
Roger Robbins
5/17/2006
Table of Contents

Purpose
The UTD Clean Room has purchased a new high performance optical Microscope from Leica, Inc. It is a manually operated microscope and is called the INM100. This document, gives brief operating instructions on how to physically operate our particular microscope. General technical background information on how a microscope works is contained in a lengthy document produced by Florida State University and can be found on their web site at http://micro.magnet.fsu.edu/primer/.
Introduction
The INM100 is a semi-automatic, high-performance optical microscope designed for the Semiconductor Industry. It is equipped with a nominal CCD camera and a computer system to capture the images. There is a large screen LCD monitor in the table with which to view and capture the images, and a camera output LCD screen mounted on the wall behind the large screen. The microscope is specially configured with an Ultra Violet Fluorescent module that will enable it to produce pseudo self-illuminated (fluorescent) images of certain organic films that may be found on samples such as photoresist, etc. The microscope is somewhat complicated with many manual knobs to turn, some combinations of which will produce total darkness through the lenses. Therefore this document should serve as a solace of understanding in the midst of confusion and perhaps darkness.
Microscope Description
Labeled photos of both sides of the microscope are shown in Figures 1 and 2. It operates basically like this: you look through the eyepieces to see a magnified image of your sample placed under the objective lenses on a manual mechanical stage illuminated by a mercury lamp at the rear of the microscope stand. This microscope utilizes reflected light only, compared to the INM200 microscope which can use transmitted light also (good for photomask viewing).
Focus is achieved by rotating the stage focus knob and moving the stage with the manual drive knobs at the lower right side of the stage. The light intensity is controlled by the thumbwheel on the front face of the microscope below the stage. The other controls manage the more subtle capabilities of the microscope and will be described individually below.

Figure 1. Right side of Microscope

Figure 2. Left side of microscope
General Microscope Operation
Startup
Before turning on the microscope, make sure that there is no sample on the stage that will collide with the objective lenses. Find the power switch on the lower right rear of the main body of the microscope and flip it on, (Figure 1). This powers up the standard mercury illumination lamp for normal viewing and also powers on the electronics that drive the microscope.
Stage Positioning
The manual stage is positioned by turning the knobs under the right front side of the stage table. The gear-engagement lever arm should be pushed away from [to the side of] the positioning knobs to engage the knob gears with the stage. The lever arm pushed the other way enables easy and rapid manual positioning of the stage (just by pushing it), to an approximation of the intended location. The upper knob moves the stage in and out relative to the operator (Y-Axis). The lower knob moves the stage to the left and right (X-Axis). The knob positioning is very precise and fine, allowing positioning of very small objects into the field of view.
With the sample in place on the stage, drive the stage manually to position the sample under the active objective lens. Make sure that there will be no collision between the objective lens and the sample – never force anything.

Figure 3. Stage drive engagement lever positions: Right – freewheeling, Left – Gears engaged.
To avoid collisions between the sample and the lenses lower the stage using the focus knobs to clear the lenses by a large margin. The objectives are mounted on a motorized rotating “nose-piece” that can be rotated to select the objective lens giving an appropriate magnification. The higher the magnification, the closer the lens comes to the sample. The lenses and nose-piece are designed so that when the nosepiece rotates to a new magnification, the image is still in focus. Therefore the operator must start with the lowest magnification (widest separation between lens and sample), and focus. After the low power focus is accomplished, the lenses may be rotated to higher power using the lens selector buttons just to the rear of the left focus knob, Figure 3. These two (red) buttons electronically rotate the nosepiece to change objectives: The top button rotates the nosepiece from low power to high power and the bottom button rotates the nosepiece in the opposite direction.

Figure 4. Focus knob and (Red) objective change buttons on the left side of the microscope.
WARNING NOTE:
One should never rotate the lenses from the lowest power to the highest power (backwards). The reason for this is that the lower power lens has a large depth of focus, making it difficult for the operator to achieve exact focus, but the higher power lens has much less depth of focus, and requires a much closer sample-lens separation. If the operator has set an imprecise focus separation on low power, the high power lens could rotate into the sample, crushing it and perhaps damaging the lens or rotator as well.
Lamp Power
Since our normal eyesight has great difficulty seeing anything in total darkness, we need light to illuminate the expected sample. The Mercury vapor lamp for standard viewing is located at the upper rear of the microscope body. It will light up when we turn on the microscope. Its intensity is controlled by the little horizontal thumbwheel control knob located on the front face of the lower microscope body under the stage. Rotate it to a numerical value of about 5 to 8 for a nominally illuminated field.

Figure 5. Mercury lamp power thumbwheel – horizontal orientation.
Filter Selection
The last control over brightness is the light input filter pushrods as shown in Figure 6. These rods rotate filters into the lamp input path, thus controlling the amount of light that passes through as well as selecting the light wavelengths input to the microscope optics. By the way, these are the pushrods located on the upper right rear of the microscope body as seen in Figure 1.

Figure 6. Light input Filter Selection pushrods.
There are three filters in this bank. The rear push-rod with no plastic ID cap is empty. The next push-rod is labeled “Diff.” (diffuser filter). This filter is merely a frosted glass lens that insures that the illumination field is uniform in brightness. It will reduce the light intensity by something like 20%. The next push-rod controls the “DLF,” or Daylight Filter that compensates for the blue shift of the Mercury arc lamp source spectrum. Inserting this filter will make the image appear slightly redder than the color without any filter. The front push-rod inserts the Blue Filter labeled “BG 20,” that
“accentuates the borders of the spectrum,” according to the instruction manual. It makes the image appear with a bluish hue.
Viewing
There are many ways of viewing your sample with this microscope: (1) Look at it with the naked eye to see approximately where your pattern of interest is in relation to the substrate so you can position it under the objectives in the vicinity of what you want to see, (2) Bright field illumination (normal magnified view), (3) Dark Field illumination (shallow angle illumination which highlights topology so that the edges of the pattern light up and the flat areas appear dark, (4) Interference contrast illumination where the contrast is enhanced or diminished by splitting the input light into two coaxial beams and then slightly adjusting the phase of one causing an interference color to appear over the image thus increasing its contrast, (5) Fluorescence illumination with short wavelength light (black light) causing an organic material in the substrate to fluoresce (give off its own light by down converting the incident high energy waves to lower wavelength (red emission), and (6) Combinations of the above. All of these methods except the naked eye will also produce an image on the LCD display on the wall and on the large screen image capture software via the CCD video camera in the tower above the microscope body.
In any case look through the eyepieces to see the sample, while driving the stage around with the right hand to locate your area of interest and adjusting the focus as necessary with the left hand – you don’t have to do anything with your feet.
Focus
Focusing the image involves moving the stage up and down until the image is sharp – in focus. The Z-axis of the stage (focus) is driven by the dual knob on both sides of the microscope base, Figure 4. The focus knob set has three functions: 1) coarse focus, 2) fine focus, and 3) max stage height lock to protect samples and lenses from collisions. The coarse focus is controlled by rotating the large diameter knob section. This allows the stage height to be moved rapidly over long distances. The fine focus knob is the smaller diameter knob that turns inside of the larger diameter coarse portion of the knob.
This is the tricky part of fine focus. If the fine focus knob section is pushed (coaxially) to the left, one rotation equals 80 microns of z-axis movement. If it is pushed to the right, one rotation equals 20 microns of stage z-movement. This extra fine positioning of the z-axis of the stage enables better control of focus for high magnification conditions.
The black ring on the right side of the microscope next to the body at the focus knob is the stage z-height lock. This prevents the stage from moving too high and causing collisions with the lenses as they are rotated. This is used in cases where the samples are thick and the stage needs to be low in order to achieve the proper focus gap between the sample and the lens. If someone then wants to look at a thin sample, the stage may not drive high enough to achieve focus. This will require the black ring about the focus knobs to be loosened and the focus knob rotated to achieve focus with a high magnification lens and then re-locked. This however puts the stage at risk from the next user who may have a thick sample. If one fails to take care in adjusting the stage for proper focus, the stage and sample may collide, causing breakage of either the sample or the lens or both.
The standard focus lock height is that which achieves proper high magnification focus for a 4 inch wafer, since most users have samples about the thickness of a 4 inch wafer. This setting results in a thin gap between the lens and the stage, so if you have a thick sample, please take extra care with the system by lowering the stage, inserting your sample, setting the objective lens to a low power away from your sample, then moving the sample under the lens, and carefully bringing it into focus. If you are viewing multiple thick samples, you might want to reset the upper bound of the stage travel to prevent
inadvertent collisions.
NOTE: if the stage does not go high enough to focus on your sample, DO NOT FORCE THE FOCUS KNOB, merely adjust the top limit setting with the right side black ring.
The black ring on the left side of the microscope body under the stage (Figure 4) is the friction adjustment ring. The friction is set by rotating the ring counterclockwise firmly to add friction to the vertical movement of the stage so that the stage will not just sink to the table level under its own weight. The optimum setting is a light friction that just prevents the stage from sinking.
Bright field imaging is the normal illumination mode for general purpose viewing. This mode lights up the entire field with bright light so you can see your sample as if from directly overhead. The bright field mode is selected by rotating the lower thumbwheel of the contrast function selection turret until it is latched into the “BF” detent. See Figure 7. The brightness of this mode is generally set by the lamp control thumbwheel on the front face of the microscope below the stage.
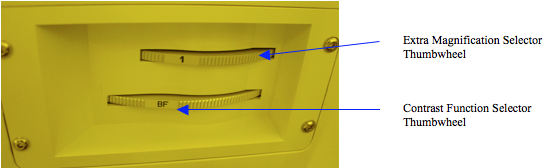
Figure 7. Bright Field “BF” selection position in the Contrast Function Selection thumbwheel.
Dark Field imaging is a technique that employs shallow angle illumination to darken flat areas but highlight edges and projections in the image field. It is especially effective in illuminating edge steps in the pattern or particles on the surface of your sample. It is selected by rotating the contrast function selector thumbwheel to the detent that shows “DF” on the flat of the thumbwheel, Figure 7. Usually this technique also requires that the illumination intensity be increased – either opening source apertures or simply increasing the lamp intensity. However this microscope has an automatic aperture control which will attempt to compensate for the dark field darkness by opening the aperture.
Condenser Aperture Contrast Control
The condenser aperture has a subtle effect on the contrast of the image. By changing the size of this aperture, the illumination cone projected into the objective lens is changed. This affects the brightness, edge contrast, and resolution of the image. The effective operating range is from 60% to 90% open. Opening the aperture too much produces glare in the image and some loss of resolution. Closing the aperture too much causes darkness and loss of resolution.
Physical control of the aperture is normally automatic (green light off), whereby the aperture is adjusted to optimum preset sizes according to the objective lens selected. The automation even adjusts for bright field and dark field illumination automatically. If your sample requires a different setting, just toggle the green light switch on and adjust the aperture size with the vertical thumbwheel located just under the green light switch. See Figure 8.

Figure 8. Details of illumination aperture control. Green light out indicates that aperture size is under automatic control according to objective lens selection. If light switch is toggled on, then manual control with the vertical thumbwheel is in effect.
This is a technique of introducing color hues by light wave interference to increase the contrast in the viewed image. Basically, the illumination light is split into two paths; one impinges on the sample and the other bypasses it and then the two beams are recombined, producing an interference effect much like the Michelson Interferometer Principle. The result is a coloration of the image as various wavelengths of light reach constructive and destructive interference because of topological steps in the sample which cause light-path length differences. Along with this coloration contrast, an additional
3-dimension like shadow effect appears that helps to bring out features of the specimen in greater contrast.
This function is selected by rotating the contrast function selection thumbwheel, Figure 7, until it seats in a detent where the “DIC” label, (Differential Interference Contrast) is showing. In addition, the DIC splitting prism thumbwheel setting, (Figure 9) must correspond to the proper objective lens. See Table 1. Once selected, the fine adjustment knob (Figure 9) allows optimization of contrast by slightly changing the interfering light beam path length.

Figure 9. Wollaston prism selection thumbwheel for Interference Contrast. This setting must correspond to the selected objective lens to produce proper contrast. The fine adjustment knob optimizes contrast by adjusting the interfering beam path length.

Table 1. Interference contrast prism selection vs objective Lens
Special Microscope Operation
Fluorescence Imaging
This microscope is fitted with a fluorescence imaging system to allow detection of organics on substrates that will fluoresce from excitation by the microscope illumination. This technique works in the following manner. Extremely bright light from a short-arc mercury lamp is directed to a “filter cube” in the body of the microscope. This filter cube filters the incoming light and passes a fixed band of excitation wavelengths through the objective to illuminate the sample. The short wavelength, high energy light then excites the molecules that will fluoresce, which then relax and emit a longer wavelength, lower energy fluorescent light. This longer wavelength must be separated from the exciting wavelength to prevent “contamination” of the signal with illumination background light. This is done in the same filter cube by a dichroic mirror which passes the fluorescent light but reflects the incoming illumination light. Because the filtering action of the dichroic mirror is incomplete, there is an additional bandpass filter on the exit port of the filter cube that sharply selects the wavelength of the fluorescent material that is finally directed to the viewing lenses of the microscope.
Now, after having read the above paragraph, it becomes apparent that this is a very specific light selection technique that must be matched to the fluorescent material you intend to examine. Fortunately, most fluorescent materials emit in either the red or green region of the optical spectrum and we have filter cubes that match both of these wavelength regions. The fluorescence mode is selected by rotating the Contrast Function Selection thumbwheel, Figure 7, to the “FL” position. Currently we have the red filter cube installed. If you need the green one, notify appropriate staff and we can physically
exchange filter cubes for your application.
There is another requirement for fluorescence illumination, and that is an extremely bright source lamp. This is provided as a separate lamp added to the rear of the microscope upper body. It is controlled by an external power supply, as shown in Figure 10. To power on the Fluorescence lamp, just turn on the Power switch. It will take a number of seconds to warm up and reach full intensity. When the lamp is at full intensity, switch from the standard lamp to the Fluorescence lamp by pulling the lever out. This rotates a deflection mirror and switches lamp sources.

Figure 10. Fluorescence Illumination lamp is on the left; power supply on right. Note that the life of this bulb is only 200 hours and it costs $200 to replace. Therefore, you must turn it OFF when you are finished.
Occasionally, you might want to have a little extra magnification to show an up-close image of a specific aspect of your sample. This can be done via the variable magnification transfer lenses which adjust the microscope internal magnification. The extra magnification is set by the upper thumbwheel in the contrast selection module as shown in Figure 7. There are 3 magnification lenses in this section of 1X, 1.25X and 1.6X. To calculate the total magnification, multiply the objective lens magnification by the extra magnification factor and then by the eyepiece magnification. For example if you were using the 100X objective and the 1.25X transfer lens for extra magnification, and viewing your sample through the standard 10X eyepieces, you would have 100×1.25×10=1250X total magnification.
In addition to this, there is a transfer lens between the microscope and the camera, Figure 11. This lens will continuously add magnification from 0.3X to 1.6X to the total microscope magnification as you rotate the body of the transfer lens tube. However, there comes a point that extra magnification just produces loss of detail in your image, so be cautious about how you utilize this feature.

Figure 11. Variable Magnification transfer lens.
Image Capture Software
This microscope has a rudimentary video image capture system that allows us to capture an image onto a flash drive memory stick. The resolution conforms to the standard NTSC video format – not really appropriate for high resolution microscopy. If you need publishable images use the INM200 microscope and its more comprehensive software.
The basic procedure to capture an image, starting from a dormant computer screen is as follows:
- Activate the Windows system by wiggling the mouse and clicking on the “Computer” icon.
- Click on the “FBSpectrum” icon in the system tray to bring up the image capture software. This icon looks like a clear box with a big, stylized red “S”
inside.
- A small window will appear with the microscope image displayed. The program name in the upper line of the window will be “FlashBus Spectrum FBG
640×480”
- To capture an image, follow the steps:
- Click “Grab” in the upper right end of the tool tray.
- This saves the image and displays the static capture image. This is the place to do any editing of the image if necessary. However the editing
tools are quite rudimentary.
- This saves the image and displays the static capture image. This is the place to do any editing of the image if necessary. However the editing
- Click “File” and find your memory stick under the “My Computer” option
- Locate your target file folder on your memory stick and open it
- Type in the name you want to give the file
- Click “Save”
- Click “Grab” in the upper right end of the tool tray.
- To return the screen to the “Live” mode, click the “Live” icon in the upper right tool tray.
Conclusion
This microscope is a high quality manual microscope for general purpose examination of your samples. It has very high magnification capabilities and also has a Fluorescence imaging capability, as well as the full standard microscope features. The scope is complex enough that incompatible feature selections may produce no image at all. This manual is intended to give you a basic understanding of the many features of the system so that you can successfully obtain revealing images of your samples. Because of this complexity, however, we would like you to be trained by the clean room staff on this microscope before actually using it. With this training and this manual, you should be able to utilize the full capability of this microscope to reveal the secrets of your samples.
Appendix A*
Suggestions from the Manufacturer
Servicing and Maintenance
- From time to time clean the outer surfaces and the contamination shield with a soft, lint-free cloth and distilled water with Isopropyl Alcohol in a 1:1 ratio.
- Accessible optics should be cleaned with a dry cotton swab (Q-Tip) or lint-free cloth.
- If the instruments and their accessories are still not clean, do not proceed with further measures. Please call the regional office of Leica for further instructions.
The Most Frequent Faults in Microscopy
Before you start using the microscope, ask yourself the following questions:
- Is the illumination correctly set?
- Is the lamp centered & aperture diaphragm correctly set for the selected objective?
- Be sure that no filters are in the optical light path that do not belong there.
- Is the revolving nosepiece correctly engaged (locked in position)?
- Is the binocular tube set for your interpupillary distance and are the focusing eye lenses of the eyepiece set correctly?
- Is the optical system clean?
Uneven Illumination
This can be caused by various errors. To begin with, check to see whether the revolving nosepiece and aperture diaphragm are in the correct position. If not, switch the microscope off and back on again and check for changes. Remove all filters in the optical path which could cause vignetting in the image. Check to see if the beam splitter of the Ergotube is in the correct position (locked in place). Check the lamp alignment.
Flat Images
Defective objectives either produce no images at all or the images are flat or move when focused through. It is often the case that front lens is damaged, although the spring mount offers a high degree of protection. Such objectives must be returned to the factory or to your local agency. Do-it-yourself repairs usually compound the defect. Dirty front lenses, however, are far more frequently seen. This should always be the first suspicion when the image lacks contrast. Finger prints and dust should be removed with a soft cloth. Resistant dirt should be removed with DI water. Occasionally the eye lenses of the eyepieces should be cleaned. They are often covered with oils from eyelashes.
Un-sharp Patches in the Microscopic Image
Un-sharp patches which remain stationary when the sample is moved, are usually caused by dust, etc. on lenses and other internal optical faces. The precise location can be seen when the eyepiece, condenser, deflecting mirror, lamp condenser, filter, etc. are rotated or moved. With some experience it is possible to determine where the dust is located by observing how the dust patches move or not with the moving elements. Here, too, cleaning should be carried out with a piece of soft rag or a soft brush.
Unnatural Contrast
The aperture diaphragm contributes to the resolution and contrast. An incorrectly set aperture can result in a too flat or too contrasty image with correspondingly reduced resolution. The correct aperture should therefore always be ensured. Never adjust brightness with the aperture diaphragm.
Lack of Fine Details because useful Magnification has been Exceeded
Excessive secondary magnification, e.g. using the magnification changer at highest magnification as standard setting, may produce “empty” magnification. The image can then lack fine details.
*Taken from the Leica Microsystems Operation Manual, Version 1.4/10.2003
Appendix B*
Software Definitions
A portion of the FlashBus Spectrum software documentation has been included here as a reference, however all of this information is available from the help file on the computer.





Appendix C*
Video Camera Operation Manual
(For Staff Only)
Reference Material for Video Camera
*Included for staff debug operations.




